Екатерина Александровна Николаева
Д.м.н., гл. науч. сотр. отд. психоневрологии
и наследственных заболеваний с нарушением психики ФГБУ «МНИИ
педиатрии и детской хирургии» МЗ РФ
Общие сведения о митохондриальных заболеваниях
Митохондриальные болезни – большая группа патологических состояний, связанных с генетически детерминированными нарушениями клеточной биоэнергетики. Заболевания отличаются тяжестью клинических проявлений, прогрессирующим течением, ограничением продолжительности жизни пациентов и вносят существенный вклад в показатели инвалидности и смертности как детей, так и взрослых, что определяет актуальность изучения данной патологии.
Энергетический обмен человека представляет собой сложный, многоступенчатый процесс метаболических преобразований: обмен пирувата, окисление жирных кислот, метаболизм цикла Кребса, функционирование дыхательной цепи, окислительное фосфорилирование. Основные этапы энергетического обмена происходят в митохондриях и ведут к аккумуляции макроэргических соединений, что обеспечивает жизнедеятельность клетки и всего организма.
Гетерогенность митохондриальных заболеваний обусловлена многокомпонентностью и особенностью генетического кодирования ядерной и митохондриальной ДНК этапов биоэнергетического метаболизма.
Митохондрии – субклеточные структуры, одна из главных функций которых заключается в утилизации и накоплении энергии органических веществ. Уникальность этих органелл связана с наличием собственной, экстраядерной ДНК. Известно, что на митохондриальной ДНК локализованы 37 генов, кодирующих транспортные и рибосомальные РНК и 13 полипептидов дыхательной цепи. Еще в 1980-е годы было установлено, что мутации митохондриальных генов имеют тяжелые клинические последствия. Однако, если в тот период митохондриальные заболевания диагностировались у отдельных больных, в настоящее время среди взрослого населения их распространенность оценивается как 1:8 500, а носительство мутаций митохондриальной ДНК в европейской популяции достигает 1:200. Дефекты митохондриальных генов обычно представлены крупными делециями, сочетающимися с дупликациями (в родословных встречаются спорадически и не наследуются), и точковыми мутациями.
Последние наследуются с высоким риском, строго по материнской линии (митохондриальное, материнское или цитоплазматическое наследование). Напротив, отсутствует риск наследования точковой митохондриальной мутации от отца. Это связано с тем, что именно материнская яйцеклетка служит источником основного пула митохондрий для организма ребенка.
В последнее десятилетие существенно трансформировались взгляды специалистов на проблему митохондриальных болезней. Стало очевидным, что дефекты митохондриальных генов представляют лишь относительно небольшую часть рассматриваемой патологии.
Другая часть связана с дефектами генов ядерной ДНК, которые кодируют подавляющее большинство из 1500 митохондриальных протеинов. Так, ядерные гены участвуют в синтезе большинства полипептидов дыхательной цепи, белков-сборщиков субъединиц дыхательной цепи на митохондриальной мембране. Биогенез митохондрий, состояние митохондриальной мембраны также контролируются ядерными генами.
Наследование ядерно-кодируемых митохондриальных заболеваний осуществляется согласно законам Менделя: аутосомно-рецессивно, аутосомно-доминантно или сцепленно с хромосомой Х.
Среди менделирующих состояний особо выделяется группа болезней, обусловленных мутациями ядерных генов, отвечающих за репликацию и стабильность митохондриальной ДНК (например, гены POLG1, POLG2, Twinkle и др.). Несмотря на ядерное наследование (аутосомно-рецессивное или доминантное), для указанных состояний характерно наличие множественных делеций или деплеции (снижение количества) митохондриальной ДНК. В связи с этим заболевания длительное время рассматривались как митохондриально наследуемые, что противоречило данным генеалогического анализа. Позднее было доказано их ядерное происхождение.
По мере изучения митохондриальных болезней коренным образом изменилось отношение клиницистов к этой патологии. Первоначально заболевания преимущественно именовались «митохондриальной миопатией», несколько позже – «митохондриальной энцефаломиопатией». Однако наблюдения показали, что эти термины совершенно не отражают развивающийся комплекс клинической симптоматики с характерным полисистемным поражением. Нервная система и скелетные мышцы страдают в первую очередь, что обусловлено наибольшей энергозависимостью клеток. Тем не менее у большей части пациентов в патологический процесс вовлекаются внутренние органы, при этом наиболее часто поражаются сердечно-сосудистая и эндокринная системы. Клинические признаки у больных встречаются в различных сочетаниях. Это привело к описанию большого числа митохондриальных синдромов, в основном с акронимными наименованиями, что не всегда оправданно, так как не отражает генез заболевания и создает трудность для дифференциальной диагностики и генетического консультирования. Лишь при отдельных формах может наблюдаться изолированное поражение определенной ткани или органа (например, при оптической нейропатии Лебера, митохондриально наследуемом сахарном диабете), однако нередко и в этих случаях по мере прогрессирования болезни манифестируют признаки миопатии, сердечно-сосудистых и других расстройств.
Поражение сердца при митохондриальных заболеваниях
Согласно мнению разных авторов, сердечно-сосудистые нарушения встречаются приблизительно у 25 % пациентов с митохондриальными болезнями (у больных раннего возраста – в 40 % случаев). Они имеют прогрессирующий характер, проявляются поражением миокарда или проводящей системы сердца, могут встречаться в виде изолированных нарушений (например, изолированная кардиомиопатия), но чаще сочетаются с другой симптоматикой, свойственной митохондриальным заболеваниям (кар диомиопатия + тугоухость, кардиомиопатия + сахарный диабет и др.). Чрезвычайно важно распознать митохондриальное происхождение заболевания, так как от этого зависят особенности ведения пациентов, тактика лечения и прогноз. Причем для правильного прогноза и медико-генетического консультирования необходимо не просто установить сам факт наличия митохондриальной патологии, а предположить дефект конкретного гена (митохондриального или ядерного) и уточнить предположение при молекулярном исследовании.
Сложность диагностики обусловлена клинико-генетическим полиморфизмом митохондриальной патологии. Одна и та же митохондриальная мутация у разных лиц (в том числе родственников) ведет к формированию разных клинических фенотипов, отличающихся не только по степени тяжести, но и по клинической симптоматике. Этот феномен обычно связывают с разным уровнем мутантной митохондриальной ДНК (гомо- или гетероплазмия, пороговый уровень) и ее распределением в тканях.
Применительно к ядерной ДНК отмечают различие клинических фенотипов, обусловленных разными мутациями одного гена. В основе этого явления, по-видимому, лежит различие локализации мутации в генном экзоне, что влечет за собой разную степень нарушения кодируемого белка. Еще больше затрудняет дифференциальную диагностику сходство клинических фенотипов, обусловленных разными ключевыми генетическими и энзимными дефектами. Так, у детей раннего возраста симптоматика болезни Ли (Leigh) или Липодобный фенотип, сопровождающийся кардиомиопатией, может развиться при нарушениях любого комплекса дыхательной цепи, дефектах митохондриальных транспортных РНК, недостаточности биосинтеза коэнзима Q10; т. е. заболевание может быть следствием различных мутаций не менее 20 генов как ядерной, так и митохондриальной ДНК. В такой ситуации, опираясь на кардинальные признаки болезни, нетрудно установить клинический диагноз.
Однако отсутствие эффективного лечения делает особенно актуальными вопросы медико-генетического консультирования и пренатальной диагностики, которые невозможно решить без точного выявления генного дефекта у конкретного больного ребенка.
Поражение сердца при дефектах митохондриальной ДНК. Сердечно-сосудистые нарушения встречаются и при делециях, и при точковых мутациях митохондриальной ДНК. Самая частая, так называемая «типичная» делеция имеет размер почти 5 тыс. пар нуклеотидов (длина молекулы митохондриальной ДНК составляет 16,5 тыс. пар нуклеотидов). Например, синдром Кернса – Сейра – одно из характерных проявлений данной делеции. Заболевание манифестирует в детском или подростковом возрасте, постепенно появляются отдельные признаки. Основной симптомокомплекс: птоз век, прогрессирующая наружная офтальмоплегия, пигментный ретинит, нарушения сердечной проводимости, мозжечковая атаксия, тугоухость, низкорослость. Поражение проводящей системы встречается почти у 90 % больных, носит прогрессирующий характер, начинаясь с дистальной блокады ножек пучка Гиса, распространяясь на атриовентрикулярный и синусовый узел с формированием полной блокады и появлением жизнеугрожающих приступов типа Морганьи – Адамса – Стокса. В 20 % случаев встречается синдром внезапной смерти. На поздней стадии болезни обычно выявляются признаки дилатационной кардиомиопатии (ДКМП).
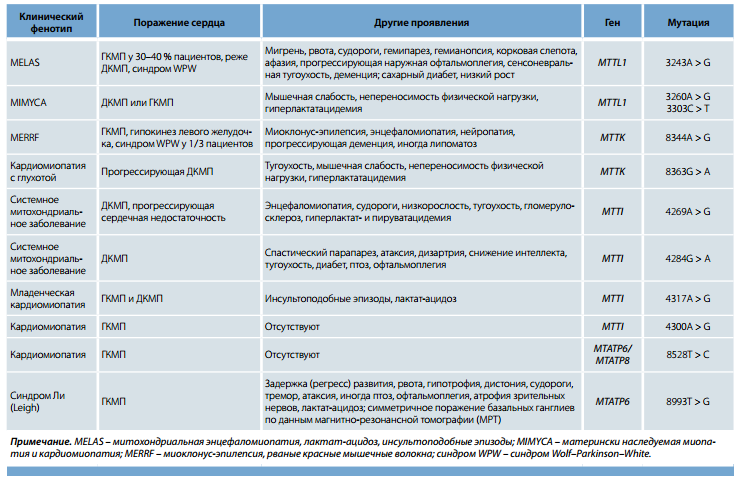
Точковых мутаций митохондриальной ДНК к настоящему моменту описано около 250, большинство из них затрагивают гены транспортных РНК, что ведет к нарушению митохондриального синтеза белка. Показано, что поражение сердца в виде кардиомиопатии диагностируется не менее чем у 20–25 % пациентов. В большей степени ассоциированы с кардиомиопатией точковые мутации генов транспортных РНК лейцина, изолейцина и лизина. Несколько реже кардиомиопатия встречается при мутациях генов транспортных РНК глицина, валина, триптофана, тирозина, генов 12S рибосомальной РНК, 1-го и 5-го комплексов дыхательной цепи и цитохрома b. Кроме того, у пациентов с дефектами указанных генов могут наблюдаться нарушения проводимости и синдром преждевременного возбуждения желудочков.
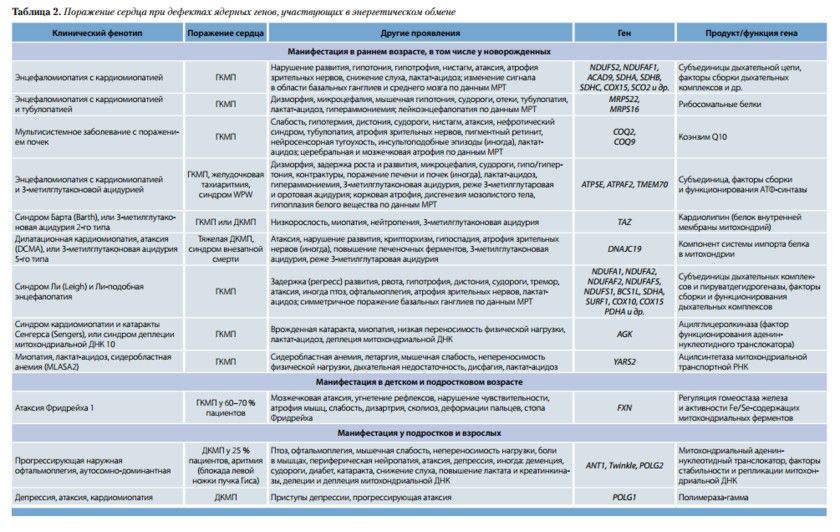
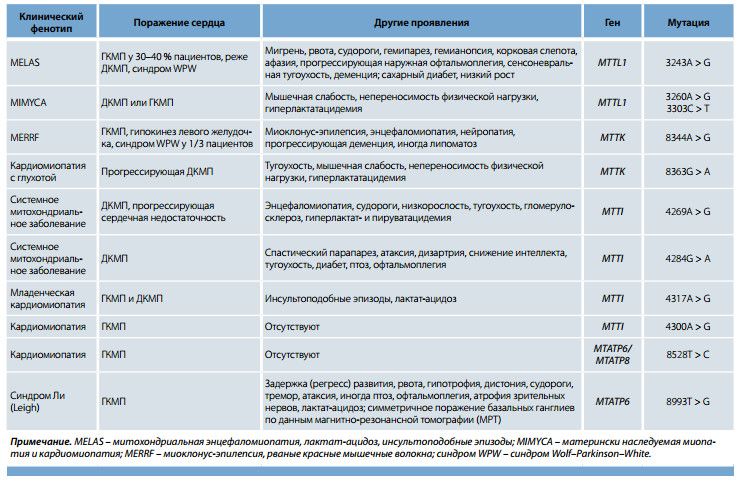
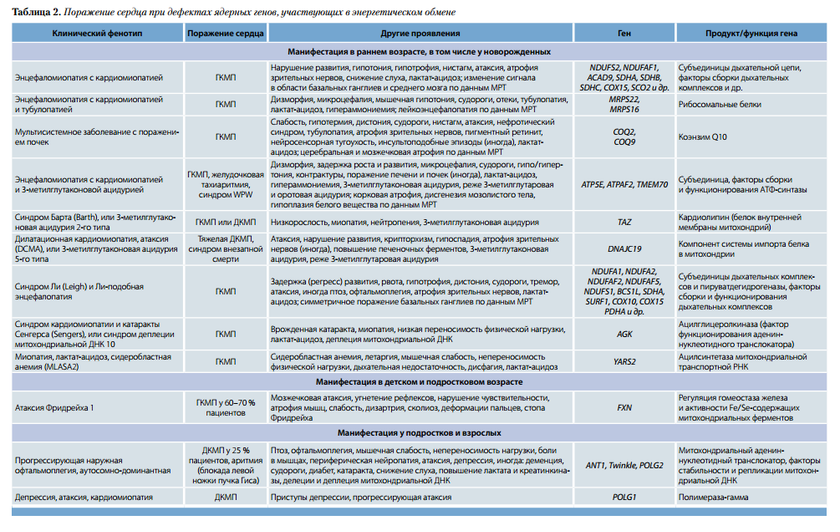
Кардиомиопатия бывает изолированной или сочетается с другими проявлениями. В табл. 1 включены наиболее часто встречающиеся точковые митохондриальные мутации, ведущие к поражению сердца.
Как показано в табл. 1, при точковых мутациях митохондриальных генов превалирует гипертрофическая кардиомиопатия (ГКМП) необструктивного типа. Дилатационный вариант встречается реже, обычно диагностируется как осложнение при прогрессировании гипертрофического варианта с расширением полостей сердца и систолической дисфункцией. Еще реже наблюдается рестриктивная кардиомиопатия с диастолической дисфункцией. Сроки манифестации сердечных нарушений различны, в некоторых случаях – ранний детский возраст.
Поражение сердца при дефектах ядерных генов, участвующих в энергетическом обмене. Большинство форм ядерно-кодиру емых митохондриальных болезней, сопровождающихся вовлечением сердца, характеризуется ранним дебютом (до 2-летнего возраста). Поражение сердца редко бывает изолированным, чаще имеет место системное заболевание. Как указано в табл. 2, в большинстве случаев развивается ГКМП, которая при отдельных формах патологии сочетается с нарушениями проводимости.
У взрослых проявления ДКМП нередко (в 25 % случаев) следуют за манифестацией птоза век и офтальмоплегии, что необходимо принимать во внимание при обследовании пациентов. Большинству представленных в табл. 2 форм свойственно аутосомно-рецессивное наследование. Рецессивно, сцепленно с хромосомой Х наследуются мутации генов NDUFA1, PDHA, TAZ; аутосомно-доминантно передаются мутации генов POLG1,
POLG2, ANT1, Twinkle, ответственные за прогрессирующую наружную офтальмоплегию у взрослых.
Диагностика и дифференциальная диагностика
Диагностика митохондриальных заболеваний основывается на комплексной оценке, прежде всего клинических и генеалогических данных. Следует обратить внимание на состояние здоровья ближайших родственников, возможное наличие у них отдельных проявлений болезни, что характерно для точковых мутаций митохондриальной ДНК. При анализе клинического фенотипа в пользу митохондриальной природы болезни свидетельствует полиорганное поражение с преимущественным вовлече нием нервной, мышечной, эндокринной, сердечно-сосудистой систем.
Важными диагностическими маркерами служат биохимические показатели: ацидоз, повышение уровня молочной и пировиноградной кислот, гипераммониемия, гипогликемия, умеренное повышение креатинфосфокиназы и трансаминаз, повышенная мочевая экскреция ряда органических кислот: 3-метилглутаконовой, 3-метилглутаровой, метилмалоновой, метаболитов цикла Кребса (фумаровой, кетоглутаровой, янтарной), а также дикарбоновых кислот. Однако необходимо иметь в виду, что изменение этих показателей может быть транзиторным.
Морфологическое исследование биоптата мышечной ткани позволяет выявить такой диагностический критерий патологии, как RRF (рваные, или шероховатые, красные мышечные волокна). При этом следует учитывать, что феномен RRF встречается не при всех формах и в то же время его можно обнаружить при заболеваниях с вторичной митохондриальной дисфункцией, например при болезнях соединительной ткани.
Безусловно, важным критерием диагностики и условием успешной дородовой диагностики (при мутациях ядерной ДНК) служит идентификация генного дефекта. На основании клинической картины и лабораторных изменений у конкретного пациента необходимо определить ген (гены), подлежащий первоочередному исследованию в молекулярно-генетической лаборатории.
Лечение митохондриальных заболеваний, сопровождающихся поражением сердца
Лечение митохондриальных заболеваний представляет очень трудную задачу, что об-условлено тяжестью нарушений митохондриальной энергетической функции и ее чрезвычайной значимостью для всего организма, отдельных органов и тканей. Лечение назначается в соответствии с патогенезом патологии, который связан с генетическим дефицитом ферментных комплексов дыхательной цепи, окислительного фосфорилирования, дефектом структурных митохондриальных белков и другими нарушениями. В результате происходит расстройство функционирования всей системы тканевого дыхания, нарушается окислительно-восстановительное состояние клеток, в митохондриях и цитоплазме накапливаются редуцирующие эквиваленты, что в конечном итоге ведет к лактат-ацидозу. Стратегия лечения митохондриальных болезней заключается в повышении эффективности биологических процессов тканевого дыхания и окислительного фосфорилирования.
Опыт многих клиницистов свидетельствует, что к настоящему моменту эффективное лечение митохондриальных болезней не разработано. В то же время для облегчения состояния и улучшения качества жизни пациентов назначается комплексная терапия, основным компонентом которой служит группа препаратов, стимулирующих перенос электронов в дыхательной цепи. Дополнительно больным назначают витамины группы В (никотинамид, рибофлавин), витамины Е и С, карнитин и др.
Сочетанное назначение энерготропных препаратов получило ироничное наименование – «митохондриальный коктейль». В то же время многими работами подчеркивается крайне низкая эффективность монотерапии.
Среди лекарственных средств – переносчиков электронов особое значение имеют коэнзим Q10 и цитохром С. Коэнзим Q10 (убихинон) играет важную роль в клеточном дыхании, обеспечивая транспорт электронов от дыхательных комплексов I и II к комплексу III. Его дополнительное введение в организм повышает активность дыхательной цепи митохондрий. Основанием для назначения коэнзима Q10 являются также данные, свидетельствующие о низком содержании убихинона в сыворотке крови и митохондриях скелетных мышц у больных с митохондриальными болезнями в связи с нарушением его эндогенного синтеза и реактивации дефектными митохондриями.
Цитохром С – одна из ключевых субстанций дыхательной цепи митохондрий, принимающая участие в переносе электронов от комплекса III к комплексу IV – цитохром С-оксидазе. Цитохром С представля-ет собой протеин, который синтезируется в цитоплазме, транспортируется в митохондрии и расположен на внутренней митохондриальной мембране в комплексе с фосфолипидами.
Цитохромы в качестве простетической группы содержат гем, в состав которого входит атом железа. Переход атома железа из двухвалентного в трехвалентное состояние (восстановленное или окисленное) и обратно обеспечивает акцепцию и передачу электронов. Окисление цитохрома С сопровождается созданием мембранного протонного потенциала, который используется клеткой для синтеза АТФ (окислительное фосфорилирование).
Препарат цитохрома С вырабатывается из ткани сердца крупного рогатого скота. В эксперименте на моделях сердечной недостаточности, ишемии миокарда, метаболического ацидоза, острого отравления окисью
углерода и др. доказано выраженное антигипоксическое действие препарата, активация окислительно-восстанови тель ных процессов в тканях. Показано, что препарат быстро всасывается и проникает в клетку. Под нашим наблюдением находилась большая группа пациентов с разными формами митохондриальной патологии. Среди них следует выделить больных с синдромами Кернса – Сейра, MELAS и Барта.
Синдром Кернса – Сейра был диагностирован у 19 детей, 12 из них была проведена моле кулярно-генетическая диагностика (Медико-генетический центр РАМН). У 10 пациентов идентифицирована типичная крупная делеция митохондриальной ДНК, у 2 в лей коцитах периферической крови делеция отсутствовала, что не исключает данный диагноз. У всех детей были выявлены прогрессирующие нарушения проводящей системы сердца. Больные получали комп лексное энерготропное лечение, в качестве одного из стимуляторов переноса электронов назначался цитохром С в дозе 4 мл в / м № 10 с повторением курса через 3–4 мес.
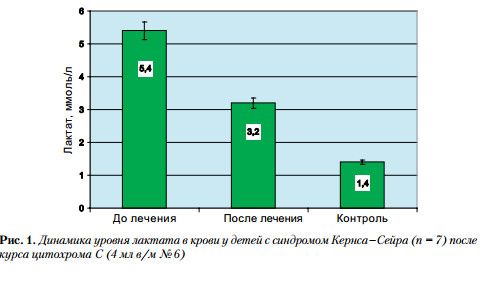
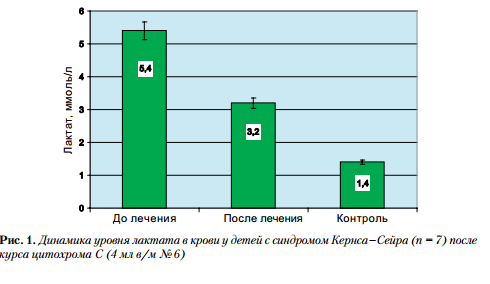
Выше подчеркивалось, что тяжесть митохондриальной патологии вынуждает использовать комплекс энерготропных средств для коррекции нарушенных процессов тканевого дыхания. Это создает сложность определения эффективности отдельных препаратов. В то же время нам удалось оценить влияние цитохрома С в коротком 6-дневном курсе монотерапии (по 4 мл в / м № 6) у 7 детей с синдромом Кернса – Сейра и гиперлактатацидемией. Было продемонстрировано убедительное снижение уровня лактата в крови с 5,4 ± 0,32 до 3,2 ± 0,16 ммоль / л, хотя нормализации уровня достигнуто не было (рис. 1).
Оценка эффективности длительного (12–14 мес) лечения с применением цитохрома С показала заметное улучшение в общем состоянии и самочувствии больных детей (n = 14): уменьшение утомляемости, мышечной слабости, моторной неловкости, стабилизация состояния сердечно-сосудистой системы, уменьшение содержания лактата в крови c нормализацией уровня более чем в 25 % случаев (у 4 пациентов). Однако при более длительном (более 3 лет) катамнестическом наблюдении за 8 больнымибыло выявлено медленное прогрессирование заболевания с нарастанием поражения проводящей системы сердца и сократительной функции миокарда по типу необструктивной симмет ричной ГКМП.
К 17-летнему возрасту у 5 детей постепенно сформировалась атриовентрикулярная блокада, что требовало своевременной установки пейсмейкера для поддержания нормального сердечного ритма. При обследовании 8 пациентов с синдромом MELAS изменения сердца в виде ГКМП были выявлены в 3 случаях. Степень гипертрофии была умеренной. Тяжесть состояния больных определялась перенесенными инсультоподобными эпизодами, повторными приступами лактатацидоза, мигренеподобной головной боли.
Длительные курсы энерготропной терапии (коэнзим Q10, цитохром С, аргинин и др.) позволили добиться улучшения самочувствия, снижения частоты и тяжести мигренозных приступов, предупреждения инсультоподобных состояний на протяжении 3-летнего наблюдения.
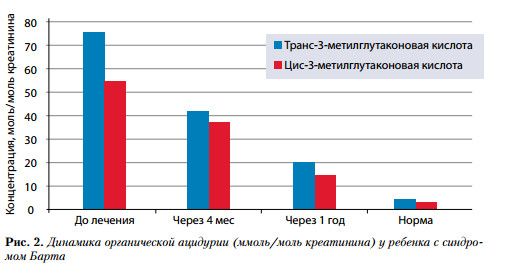
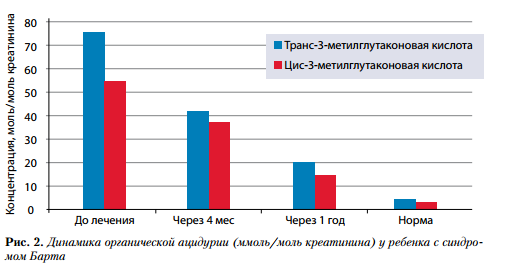
Выраженная эффективность была отмечена при лечении ребенка с синдромом Барта. В возрасте 3 лет у мальчика были выявлены низкорослость, миопатический синдром (слабость, гипотония, нарушение походки), необструктивная симметричная ГКМП (толщина задней стенки левого желудочка 11 мм, межжелудочковой перегородки – 11,8 мм при норме 5,5 мм), транзиторная нейтропения (количество ней-трофилов 880 в 1 мкл при норме 2300), очень высокая (более чем в 30 раз выше нормы) почечная экскреция 3-метилглутаконовой кислоты. В комплексе лечения ребенка основными препаратами были цитохром С (4 курса в год) и левокарнитин
(от 500 до 1000 мг / сут длительно). Благоприятный эффект проявился ликвидацией миопатического синдрома к возрасту 7–8 лет, полной нормализацией эхокардиографических показателей и параметров длины тела к возрасту 9–10 и 12 лет соответственно. Заметно снизилась, но осталась повышенной экскреция 3-метилглутаконовой кислоты (рис. 2).
Заключение
Поражение сердца вносит существенный негативный вклад в состояние пациентов с митохондриальными заболеваниями, в значительной степени определяя уровень летальности и продолжительность жизни.
Проблема лечения этих тяжелых заболеваний далека от разрешения. Патогенетическая терапия сердечно-сосудистых нарушений митохондриального происхождения направлена прежде всего на коррекцию тканевого обмена, повышение уровня окислительного фосфорилирования. В комплексе энерготропных препаратов важное место принадлежит цитохрому С – антигипоксическому средству, активатору транспорта электронов в дыхательной цепи. Использование этого препарата способствует стабилизации патологического процесса, а в ряде случаев ведет к значительному улучшению состояния больных.
Источник: Газета «Кардиология сегодня» №1 2014














мне кажется — немного не тот электорат здесь для таких материалов. 90% ни слова не поймут ) на графиках отсутствует р — величина погрешности исследований.
дело нужное. обидно — что информация на открытых ресурсах — это полный треш — и именно она является формирующей отношение...
увы, нужно фильтровать все… иногда и научные статьи бывают бредовыми, как например, ученый, которые опровергает существование СПИДа и вроде как даже научно его обосновывает, но изначально у него заложена логическая ошибка)))) а так то красиво писал))))